The research conducted in our lab focuses on two aspects: 1) developing quantitative models and computational methods for analyzing high-throughput data produced by emerging genomics technologies; and 2) applying innovative computational and data science approaches to study epigenetics and transcriptional regulation of gene expression in mammalian cell systems and human diseases such as cancer.
Understanding how gene expression is regulated in chromatin is a fundamental question in molecular biology. The transcription program is a major determinant of cell identity, and transcriptional regulation plays a crucial role in various biological processes and human diseases. Advanced genomics technologies, including high-throughput sequencing and single-cell and spatial omics assays, enable us to obtain massive amounts of data that measure the dynamic patterns of numerous factors and elements in the genome. These factors and elements influence chromatin states and gene regulation. We harness the power of big data and integrate computational and experimental research at the intersection of functional genomics, epigenetics, and cancer biology. Several ongoing research directions include:
1. Bioinformatics methods for emerging omics technologies
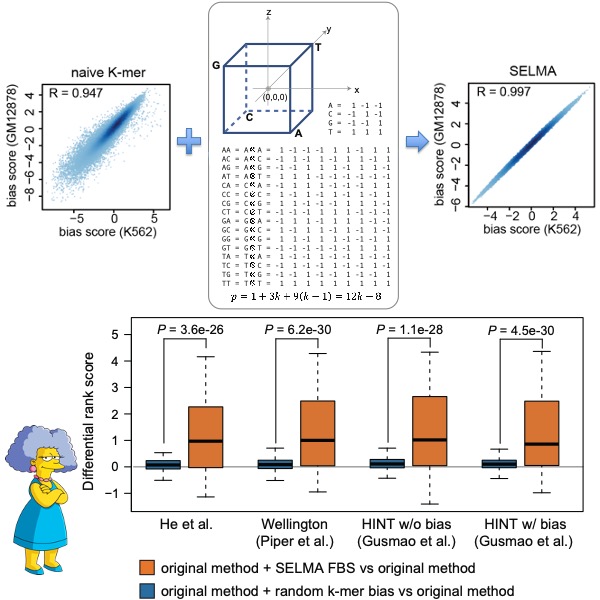
The advancement of life sciences has been significantly accelerated by new technologies. Developing innovative analytical methods is crucial for transforming high-throughput experimental data into scientific knowledge. One focus of our lab is to create innovative statistical models and algorithms for analyzing data generated from emerging genomics technologies. As a pioneer in next-generation sequencing (NGS) bioinformatics, we developed SICER (Bioinformatics 2009), one of the most widely used methods for ChIP-seq data analysis. Currently, we are developing new methods for the unbiased analysis of data from epigenomics (ATAC-seq, CUT&RUN, CUT&Tag, etc.), single-cell multi-omics, and spatial omics techniques (Nat Commun 2022). These methods aim to enhance our understanding of gene regulation.
2. Machine learning methods for regulatory factor prediction and multi-omics integration
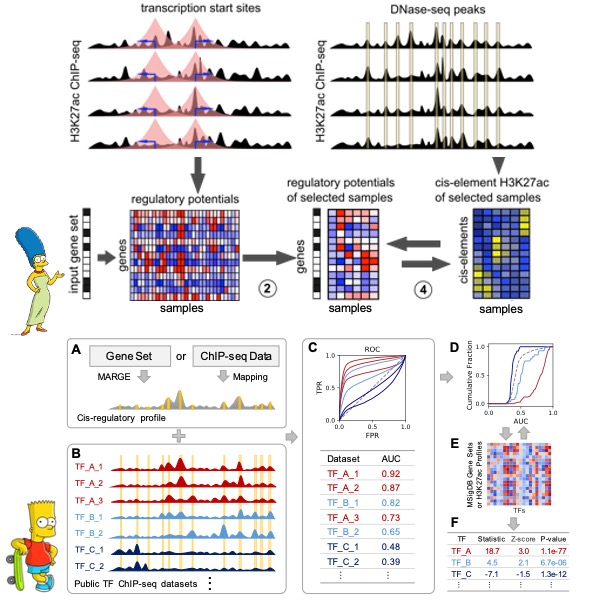
Transcriptional regulators (TRs), including transcription factors and chromatin regulators, play a key role in transcriptional regulation. Leveraging publicly available ChIP-seq data, we have developed a series of machine learning-based computational methods, including MARGE (Genome Res 2016), BART (Bioinformatics 2018), BARTweb (NAR Genom Bioinform 2021), and BART3D (Bioinformatics 2021). These tools are designed for predicting cis-regulatory profiles and functional TRs from various input data types. By integrating public omics data with the Cancer Genome Atlas (TCGA), we created BART Cancer (NAR Cancer 2021) to model transcription factor activities in TCGA cancer types. Currently, we are developing new methods tailored for single-cell multi-omics data and aim to establish a comprehensive framework using advanced machine learning techniques for data integration and TR prediction.
3. Data-inspired modeling for functional epigenetics and transcriptional condensation
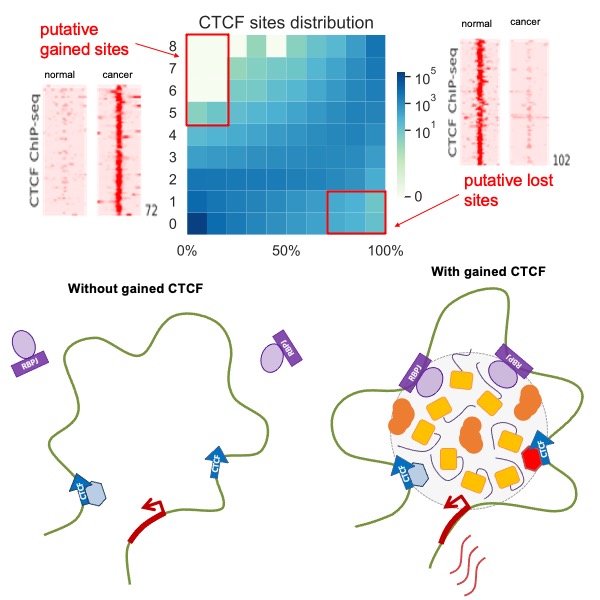
Data-driven discovery has become a new paradigm in biological research. By analyzing the massive amount of data available in the public domain, we can uncover new patterns and relationships in biological entities that often remain hidden in individual datasets. By the integrative analysis of thousands of public omics datasets, we have recently identified a cancer-specific binding pattern of CTCF, an important DNA-binding protein, and have characterized its role in facilitating oncogenic transcriptional activation (Genome Biol 2020). Inspired by the emerging evidence of liquid-liquid phase separated biomolecular condensates (e.g., Nature 2021), we develop computational models to characterize transcriptional condensates from genomics data (Nucleic Acids Res 2025). Our ultimate goal is to gain a deeper understanding of the molecular mechanisms underlying transcriptional regulation.
Research Talks
Collaborations
Computational biology is an interdisciplinary science. It cannot thrive without close collaboration among researchers from diverse backgrounds and areas of expertise. We are committed to fostering collaborations and embracing team science, working alongside experimental biologists, clinicians, statisticians, computer scientists, mathematicians, and physicists. Our research spans functional epigenetics and transcriptional regulation across various biological systems and human diseases, including T-cell immunity, malignant peripheral nerve sheath tumor (MPNST), acute myeloid leukemia (AML), T-cell acute lymphoblastic leukemia (T-ALL), colorectal cancer, coronary artery disease, development, and environmental health, etc.
Our collaborators include:
Myles Brown, Department of Medical Oncology, Dana-Farber Cancer Institute, Harvard Medical School
Danfeng Cai, Department of Biochemistry and Molecular Biology, Johns Hopkins University
Suresh Cuddapah, Department of Medicine, New York University
Anindya Dutta, Department of Genetics, University of Alabama at Birmingham
Siyuan Wang, Department of Genetics, Yale University
Hai-Hui Xue, Center for Discovery and Innovation, Hackensack University Medical Center
Zhen Yan, Center for Exercise Medicine Research, Virginia Tech
Xu Zhou, Boston Children's Hospital, Harvard Medical School
Todd W. Bauer, Division of Surgical Oncology, Department of Surgery